RMC
|
Molecular RMC simulation with FNC |
|
Summary |
This page describes the implementation of the molecular move option. Although this is still
available in the code for the CCl4 molecule and among the source
files molecular move source files for C2Cl4 and H2O
can be found and can be compiled into the code, this approach, and generally
moving more than one atom at the same time did not proved to be advisable, as
it reduced the number of tried and accepted moves significantly. In case of the
molecular moves the molecules were defined with the Fixed Neighbour Constraint,
FNC. Applying FNC for keeping molecules together and only moving just one atom
(so not applying molecular move) was successful for some small molecules as
well. Later on, when RMC_POT was created, the flexible molecules
approach was introduced, where the molecules were kept together by bonded
potential, and also only one atom is moved in the same time. But the
description below is given for historical purposes.
|
Moving one molecule instead of one or more
atom at each step |
In standard RMC, each step in
the random walk of the Metropolis algorithm consists in the move of one or more
randomly chosen atom.
This is suitable, if the molecules of the material one is trying to model are
flexible enough for such displacements to be physically acceptable.
However there are cases were the geometry of the molecules is known a priori,
furthermore, there are also cases were this geometry is not much flexible. As a
result,
- either large individual moves are allowed and the
resulting molecules are not physically acceptable,
- either the allowed individual moves are kept small
enough not to destroy the molecular geometry, but then the configuration
will find it very difficult (i.e. slow) to evolve.
The solution to get out of this situation is not to move just one atom
at a time, but the whole molecule, while keeping its geometry.
Thanks to the fixed-neighbour constraints (FNC) option, individual atoms can be
'linked' to define molecules within a configuration. Furthermore, RMC++ has
been implemented in a way that the simultaneous move of several atoms is
possible.
Note that it
does not make much sense to move randomly several individual atoms at the same
time. The result would not be different than making the several individual
moves sequentially. Multiple atoms moves only make sense if groups of
atoms are moved in some defined way.
It must be noted that in the
whole algorithm, the only step that is modified by this 'collective displacement'
is the move. Calculation of distances and checking of constraints are not
affected.
As a result, the change from 'individual atomic move' to 'molecular' move
requires only the modification of a few routines in the whole RMC++ code.
It must be noted however,
that these moves are custom made, i.e
for different molecules one might define different moves, translations along
preferred directions, rotations along preferred axes, etc... Each custom move
requires the implementation of the corresponding Move function in the code (and
hence a little bit of programming that requires some knowledge of how RMC++ is
implemented).
Some example of custom moves are shown below.
|
Examples |
C2Cl4
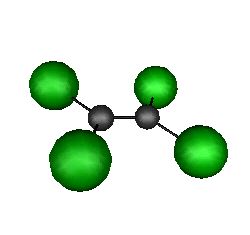
C2Cl4 is a
rigid, flat molecule for which individual atomic moves are prohibitively small
if an acceptable molecular geometry is to be kept. On the other hand, if the
whole molecule is to be moved one can define three
preferential axes for rotations and translations as shown below.
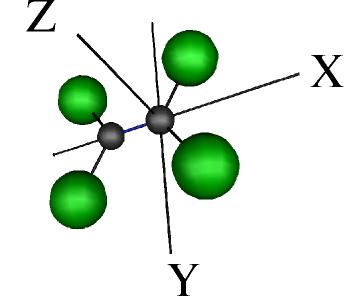
Note that the
definition of specific directions usually requires making a distinction between
atoms even of identical atomic species within the molecule. The geometry is
defined in the FNC file by setting as many as necessary FNC links between atoms
of the same molecule. The FNC settings for C2Cl4 are shown below. Note also
that the numbering of constraints defined in the FNC file are
completely independent from the atomic types that appear in the main RMC
routine for computing the partials and the weighting of the different structure
factors.
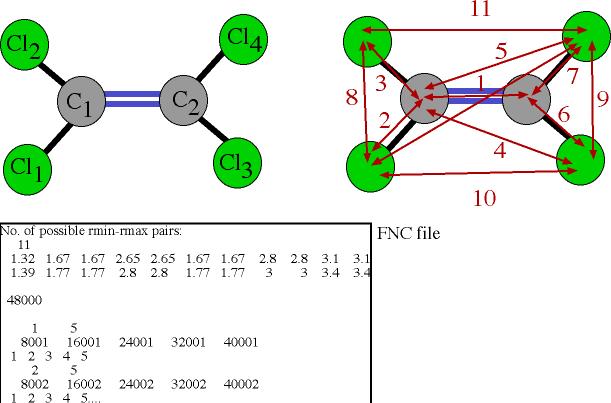
In this
particular case, some FNC constarints are
unnecessarily redundant, but the main point in the FNC file is to define the
sets of neighbours that make up a molecule. In this particular case, it is
essential that each atom has 5 neighbours. Note also that the implementation of
the molecular network in the *.cfg and in the *.fnc files must be known in order to program the custom
move.
CCl4
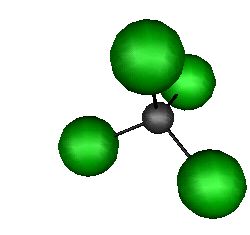
The shape of
the CCl4 molecule can be well defined by a set of FNC constraints. Depending on
the level of flexibility that one gives to the molecule, individual atomic
moves can be acceptable or not. Note that RMCA-type individual atom moves can
also be implemented within the custom molecular move (molecules have a defined
shape with some flexibility).
In the case of CCl4, one can define many specific directions, or decide that
there is no a priori preferred direction for rotations and translations,
i.e. they can be performed along random axes.
H2O
The shape of
the H2O molecule is also well defined by FNC constraints alone. The figure
below indicates what kind of flexibility some FNC ranges imply.
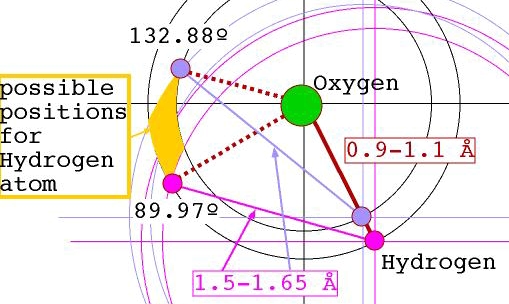
Molecular
move allows much more rigidity for the molecule, while allowing the
configuration to evolve.
Additionally, it is possible to define preferential axis of rotation and
translation for the molecular move, or, alternatively to choose randomly these
directions.
CS2
Finally, CS2
seems to be a particularly good example for applying the molecular option of
RMC++.
Indeed, this molecule has the structure of a 'rod': the three atoms are
aligned. Defining the alignment with FNC constraints alone would imply either
very small individual atomic move amplitudes, or alternatively, a very loose
alignment, as seen below.
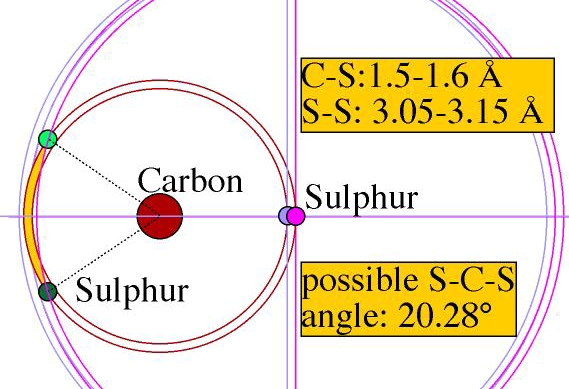
Molecular move allows both molecular rigidity and large move amplitude so that the configuration evolves.
|
Limitations |
It is clear, that while
moving whole molecules rather than single atoms solves some problems, it also
yields additional difficulties. The first obvious problem is space, i.e.
space to move whole molecules. Indeed, at the densities of most materials for
which RMC is of some use, molecules will 'bump' into each other very easily,
and that puts very strong limitations to the move amplitudes. There is no
immediate answer to this problem. Getting closer to reality by e.g.
moving several molecules at a time, is just a change of scale...
|
Example routines |
Hereafter you can find a set of custom
move routines that have been designed for CCl4, C2Cl4 and H2O.
RMC_POT can handle both atomic and molecular moves as well.
The download version on downloads page includes
the makemovecus.cpp source file which contains the custom routines for
the CCl4 molecular RMC simulation. See the manual
for details.
Additional *.cus input parameter file
The free parameters that may be needed in the
implementation of the custom move are stored in a file with file name*.cus, which is read at the start of the run. The
content of this file depends on the custom move and the reading procedure is
part of the constructor that is implemented in the makemovecus
file.
Example files to download
There are a set of custom files for molecular RMC++
available for download:
- For Windows, click here
(same text files with DOS line breaks, compressed with WinZip)
- For Unix/Linux click here
(same text files with Unix line breaks, compressed with tar and gzip)
- There is a test simulation example in the validation
suite’s CCl4_mol directory for molecular RMC for CCl4, see the download page.
Last modified 04/03/2023) by Orsolya Gereben
(comments welcome!)