
CONTRIBUTIONS
| Marco Amores: Fast microwave-assisted solid-state synthesis of lithium containing garnets and local structure characterization by total scattering (poster) |
| Mona Bahout: Stability under hydrogen and humid conditions of potential SOFC electrodes investigated by high-temperature in situ neutron powder diffraction (talk) |
| Imre Bakó: A new approach to the determination of the uncertainty in neutron diffraction experiments with isotopic substitution method (talk) |
| Rafal Babilas: Reverse Monte Carlo modeling of local atomic structure in binary Fe-based metallic glasses (poster) |
| Dominic Carter: The local structure of beta-cristobalite as a composite of low symmetry domains (talk) |
| Viviana Cristiglio: Structure investigation of molecular liquids and liquid solutions by small angle neutron scattering (talk) |
| Kaustuv Datta: Local structures of perovskite based ferroelectric solid solutions containing morphotropic phase boundary (talk) |
| Juan Du: After RMC refinement: new tools for analysing refined configurations (with case study BiFeO3) (talk) |
| William Fletcher: A Bayesian route to protein structure without crystals (talk) |
| Giles Flowitt-Hill: Local order of small organic molecules in crystals (talk) |
| Nick Funnell: RMC and the Nanoscale (talk) |
| Orsolya Gereben: Characterization of the hydrogen-bonded network in ethanol-water mixtures (talk) |
| Andrew Goodwin: RMC in magnets (talk) |
| Ilkyoung Jeong: Separation of thermal disorder from static displacements in RMC modeling (talk) |
| Andrew J. Johnston: The Atomic Structure of Pharmaceuticals in Solution: Penetrating the Blood-brain Barrier (talk) |
| Pál Jóvári: Short range order in CuZr-based metallic glasses (talk) |
| David Keen: The first 27 years of reverse Monte Carlo (talk) |
| J. Vidal Laveda: Microwave synthesis of LiFe1-xMnxPO4 nanostructures for use as positive insertion electrodes in Li-ion batteries (poster) |
| Huw Marchbank: Understanding how different synthetic procedures influence the short, medium and long range atomic arrangement in ceria (talk) |
| Serena Ada Maugeri: Modelling nanoparticles structure using PDFgui and RMCprofile (talk) |
| Sylvia McLain: Probing biomolecular structure in aqueous solution: Insights from neutron diffraction and computation (talk) |
| Bahout Mona: Stability under hydrogen and humid conditions of potential SOFC electrodes investigated by high-temperature in situ neutron powder diffraction (talk) |
| Koji Ohara: Liquid structure of the electrolyte material Li/Mg/Cs-TFSA molten salt (talk) |
| Yohei Onodera: Reverse Monte Carlo methods for superionic conductors (talk) |
| Alistar Overy: Disorder-phonon coupling in crystal-like aperiodic solids (talk) |
| Lewis Owen: Short range order in alloys (talk) |
| Luis Alberto Rodríguez Palomino: The structure factor of liquid propanol studied by polarized neutron diffraction and RMC (talk) |
| Anthony E. Phillips: Phase transitions in molecule-containing crystals (talk) |
| Helen Playford: How long is long enough? (talk) |
| Ildikó Pethes: Chemical order in chalcogenide glasses (talk) |
| Szilvia Pothoczki: Intermolecular correlations in liquid acetonitrile (talk) |
| László Pusztai: Determining the structure of molecular liquids by combining molecular dynamics and RMC (talk) |
| Vicente Sanchez Gil: N-RMC method: Reverse Monte Carlo modeling in confined systems (talk) |
| Arkadiy Simonov: 3D-∆PDF + RMC: a microscopic model of orientational frustration in an organic single crystal (talk) |
| Alan Soper: Recent developments in EPSR (talk) |
| Nicola Steinke: Atomic level insights into urea induced protein unfolding (talk) |
| László Temleitner: H-bonded and non-H bonded liquids: removing the incoherent scattering of 1H polarized neutrons (talk) |
| Janis Timoshenko: Reverse Monte Carlo/evolutionary algorithm approach for the analysis of EXAFS data from distant coordination shells of crystalline materials (talk) |
| Rupert Tscheließnig: Interfacial forces of proteins and surfaces (talk) |
| Phillip Tucciarone: NTE, spaghetti dynamics, and hydration-driven volume collapse in ZrW2O8 (talk) |
| Matt Tucker: The new XPDF beam line at Diamond (talk) |
| Peter Thygesen: Small displacements, big effect: correlated orbital disorder drives spin-glass state in Y2Mo2O7 (talk) |
| Hans Weber: Atomic structure of liquid GeTe (poster) |
ABSTRACTS
Stability in humid conditions of potential SOFC electrodes investigated by high-temperature in situ neutron powder diffraction
Mona BAHOUT1, Stevin S. Pramana2, James M. Hanlon1, Vincent Dorcet1, Ron Smith3, Serge Paofai1 and Stephen J. Skinner2
Cobalt-based double perovskite oxides possess excellent electrochemical properties correlated with their mixed ionic and electronic conductivity and are being considered as potential cathodes in H+-SOFCs. Their fast oxygen transport kinetics and the high level of oxygen vacancies they can accommodate are needed to allow the dissociation of water gas molecules and the formation of protonic defects; however the presence of structural protons in these systems has not yet been demonstrated.
In situ neutron diffraction data collected on the high-flux POLARIS diffractometer at ISIS (RAL, UK) exploited the sensitivity of neutron diffraction to light atoms to determine atomic positions and site occupancies. The double perovskites NdBaCo2-xMnxO5+δ (x = 0 and 0.5) were investigated using high temperature neutron powder diffraction in dry argon and wet atmospheres [1]. This enabled us to search for the presence of protons, monitor the oxygen vacancy formation and assess the stability and structural behaviour of NdBaCo2-xMnxO5+δ under operating conditions consistent with cathodes in H+-SOFCs.
References:[1] M. Bahout, S. S. Pramana, J. M. Hanlon, V. Dorcet, R. I. Smith, S. Paofai and S. J. Skinner, Journal of Materials Chemistry A, 3 15420 (2015)
A new approach to the determination of the uncertainty in neutron diffraction experiments with isotopic substitution method
Imre BAKÓ 1, Gábor Pálinkás , Tamás Grósz 2, Szabolcs Bálint 2, Gergely Tóth 3
Corresponding author: Imre Bakó , bako.imre@ttk.mta.hu, 003614883891,
Neutron diffraction experiment with isotopically substituted substances is a powerful approach claiming to yield unambiguous information about the local atomic structure in disordered materials. This information is expressed in the partial structure factors, and extracting them from a series of measurements requires solution of a set of linear equations that is affected by experimental errors. In this article, we suggest a method for the determination of the optimal set of H/D compositions with or without taking into account the experimental error. For the case of water, our investigations show that the selection of the isotope concentrations and the distribution of measurement time among the various samples have critical role if one wants to utilize the limited neutron beam time efficiently. It is well known that measurements of pure H2O introduce fairly large errors in the partial structure factors due to its very strong incoherent scattering. On water and methanol as examples, we investigated the propagation of random errors to the partial structure factors using partial pair-correlation functions from molecular dynamics simulation. We investigated the effect of incorrect normalisation and subtraction of multiple scattering contribution to the quality of partial structure functions. It is shown on the example of water that it is not worthwhile to measure pure H2O.
Reverse Monte Carlo modeling of local atomic structure in binary Fe-and Co-based metallic glasses
R. BABILAS1, A. Burian2, L. Hawelek3 and L. Temleitner4
The work was supported by National Science Centre under research project no.: 2011/03/D/ST8/04138.
Structure investigation of molecular liquids and liquid solutions by small angle neutron scattering
Viviana CRISTIGLIO
The second part of the talk will concern the investigation of changes in protein-protein interaction distances of a model protein/cryoprotectant system based on lysozyme/sorbitol/water liquid solution using small-angle neutron scattering at D22 instrument at ILL. Many protein drugs are either stored as frozen solutions, or converted into the solid state by freeze-drying, in order to improve the long-term stability. Aggregation is often observed after freeze-thaw or reconstitution of freeze-dried powder and the stability is no longer assured. These neutrons results demonstrate the utility of SANS methods to monitor the protein crowding at different stages of freezing and drying and the role of the carbohydrates in the protein aggregation.
The organization of the molecular structures in these liquids systems is not fully understood and a possible integration of RMC refinement in these data sets could be really helpful in data interpretation at the nanoscale range order.
A Bayesian route to protein structure without crystals
William FLETCHER
References: [1] Y. Jiao, F. H. Stillinger and S. Torquato, Phys. Rev. E, 81, 11105 (2010) [2] M. J. Cliffe, M. T. Dove, D. A. Drabold and A. L. Goodwin, Phys. Rev. Lett., 16, 2654 (2010)
RMC and the Nanoscale
Nick FUNNELL, Qiang Wang, Matt Tucker, Leigh Connor, Dermot OHare, Max Fulford, Alex Ha and Andrew Goodwin
References: [1] M. Tucker, D. Keen, M. Dove, A. Goodwin and Q. Hui, J. Phys.: Condens. Matter, 2007, 19, 335218 [2] N. Funnell, Q. Wang, L. Connor, M. Tucker, D. OHare and A. Goodwin, Nanoscale, 2014, 6, 8032
Characterization of the hydrogen-bonded network in ethanol-water mixtures
Orsolya GEREBEN
Separation of thermal disorder from static displacements in RMC modeling
Ilkyoung JEONG
Local atomic displacements and nano-scale ordering are important features of mixed-ion ferroelectric perovskite and play a key role in their macroscopic properties. For example, a formation of nano-scale polar regions in the average cubic lattice has been reported in a relaxor ferroelectric Pb(Mg1/3Nb2/3)O3 (PMN). For a structural modeling of these complex materials, reverse Monte Carlo (RMC) method is often adopted to handle both local and the average structural features simultaneously. In this talk, I will discuss how the separation of thermal disorder affects atomic structure from RMC modeling by using DISCUS [2] and RMCprofile [3] software.
References: [1] I.-K. Jeong et al., Physical Review Letters 94, 147602 (2005). [2] Matt Tucker, Stefan Norberg, Andrew Goodwin, Martin Dove, Toby White, Victor Krayzman, Igor Levin, http://www.rmcprofile.org [3] Th. Proffen and Reinhard Neder, http://discus.sourceforge.net
The Atomic Structure of Pharmaceuticals in Solution: Penetrating the Blood-brain Barrier
Andrew J. JOHNSTON, Sebastian Busch, Richard J. Gillams, Sam Callear and Sylvia E.McLain
Microwave synthesis of LiFe1-xMnxPO4 nanostructures for use as positive insertion electrodes in Li-ion batteries
J. Vidal LAVEDA1, M. Tucker2, H. Playford2 and S. A. Corr1
Here, we present two microwave-assisted synthetic approaches for the preparation of LiFe1-xMnxPO4 nanoparticles, one using commercial starting materials and other employing a new class of heterometallic alkoxide precursors. Co-location of two transition metals in these metalorganic precursors is believed to bypass the need of diffusional mixing and allow the reactions to proceed faster and at lower temperatures generating highly crystalline materials. To fully characterize and have a better understanding of the structure-property relationship of these nanocrystalline phases, we show neutron pair distribution function (PDF) analyses of these materials, which allow elucidation of the local structure, cation distribution, presence of defects and Li content. Future work involves modeling this neutron PDF data using the Reverse Monte Carlo method. Finally, we also include cycling studies in an effort to probe the relationship between the synthetic route, composition and electrochemical performance.
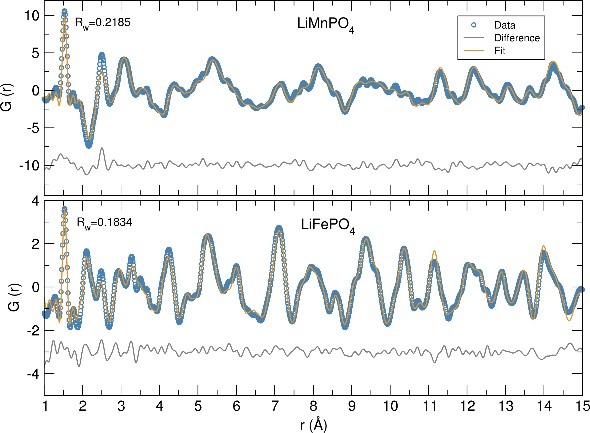
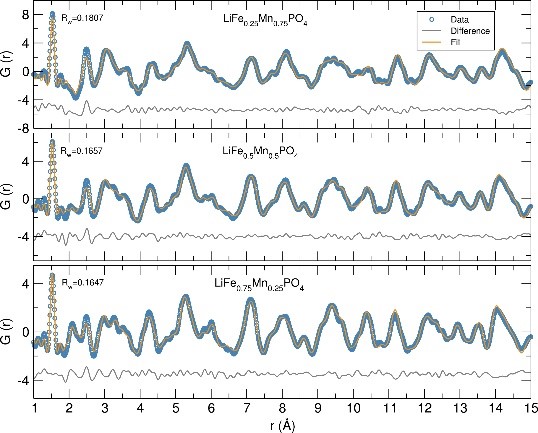
References: [1] Tarascon, Armand, Nature, 2001, 414, 359. [2] Kang, Ceder, Nature, 2009, 458, 190 [3] Aravindan, Gnanaraj, Lee, Madhavi, J. Mater. Chem. A, 2013, 1, 3518. [4] Manthiram, Murugan, Sarkar, Muraliganth, Energy Environ. Sci., 2008, 1, 621. [5] Ashton, Vidal Laveda, MacLaren, Baker, Porch, Jones, Corr, J. Mater. Chem. A, 2014, 2, 623
Understanding how different synthetic procedures influence the short, medium and long range atomic arrangement in ceria
Huw MARCHBANK
The use of both in-situ x-ray and neutron PDF techniques have been successfully used to characterise both lanthanum-doped ceria [4] allowing greater understanding of local deviations from the average structure. Similarly the use of neutron PDF techniques have been used to investigate the effect of temperature treatment of ceria based materials [5].
Ex-situ XAFS, x-ray and neutron total scattering techniques have been used to investigate the ceria in order to show how the crystalline structure varies from the short through to long range e.g. different syntheses and how these influence the defect chemistry, different techniques and their respective view of the materials. These materials were studied further by the use of RMCProfile [6] in order to combine all these methodologies to gain a thorough understanding of the crystal structure under reaction conditions.
References: [1] T. Bunluesin, R.J. Gorte and G.W. Graham, Appl. Catal., B, 1998, 15, 107-114 [2] C. Tyrsted, K.M. Ørnsberg Jensen, E. D. Bøjesen, N. Lock, M. Christensen, S. J. L. Billange and B. B. Iversen, Angew. Chem. Int. Ed., 2012, 51, 9030-9033 [3] P. Li, I.W. Chen, J. E. Penner-Hahn, and T.Y. Tien, J. Am. Ceram. Soc., 1991,74, 958967. [4] M. Coduri, M. Scavini, M. Brunellic, and P. Masalaa, Phys.Chem.Chem.Phys.,2013, 15, 8495. [5] E. Mamontov and T. Egami, J. Phys Chem. Solids, 2000, 61, 1345-1356. [6] M.G. Tucker, D.A. Keen, M.T. Dove, A.L. Goodwin, and Q. Hui, J. Phys. Condens. Matter, 2007, 19, 335218.
Probing biomolecular structure in aqueous solution: Insights from neutron diffraction and computation
Sylvia E. MCLAIN, Andrew J. Johnston, Nicola Steinke, Sebastian Busch, Luis Carlos Pardo, Christian D. Lorenz and Richard J. Gillams
Structural study of sulfide-based crystalline/glassy superionic conductors by RMC modeling
Yohei ONODERA
References: [1] F. Mizuno, A. Hayashi, K. Tadanaga, M. Tatsumisago, Adv. Mater. 17 (2005) 918. [2] A. Hayashi, K. Noi, A. Sakuda, M. Tatsumisago, Nature communications 3 (2012) 856. [3] N. Kamaya, R. Kanno et al., Nature Materials 10 (2011) 684.
The structure factor of liquid propanol studied by polarized neutron diffraction and RMC
L. A. Rodríguez PALOMINO1,2, G. J. Cuello1, A. Stunault1, J. Dawidowski2, L. Temleitner3
We present the experimental structure factor of liquid 1- and 2-propanol measured in the Polarized Hot Neutron Beam Facility (D3) at the Institut Laue Langevin (ILL, France). This neutron technique has the advantage of experimentally separating the coherent and incoherent scattering intensities. Using a linear combination of the non-spin-flip and spin-flip diffractograms, one can determine the coherent intensity, related with the coherent structure factor. We present a new procedure to perform the experimental correction using a hybrid Monte Carlo simulation code development for this kind of experiments. With this code, we evaluate the corrections by multiple scattering, attenuation and inelasticity (produced by the sample and the sample cell) as a whole and not as independent corrections, as it is usually done in the standard methods employed in neutron diffraction experiments. This hybrid simulation code is based on the combination of a modeled energy exchange and the experimental angular distribution, which are employed as input in the simulations. The good agreement observed between our simulation and the experimental results, confirm the goodness of this model. We performed RMC simulations using our corrected experimental structure factor. In doing this, we convoluted the ideal structure factor obtained from the RMC with the instrumental resolution in Q-space. We obtain a very good agreement between the simulated and experimental structure factor, which allows us to study the intermolecular structure in the simulation box.
Chemical order in chalcogenide glasses
Ildikó PETHES
Intermolecular correlations in liquid acetonitrile
Szilvia POTHOCZKI, László Pusztai
The following findings emerged from the present study: (1) Molecular Dynamics simulation is necessary to estimate the intermolecular correlations; (2) dipole-dipole correlations beyond the first coordination shell (where the molecules form typically antiparallel arrangements) show particular tendencies; (3) characteristics of partial radial distribution functions and distance-dependent orientational correlation functions exhibit well-recognizable structural features far beyond the first coordination shell.
References: [1] Sz. Pothoczki, L. Temleitner, P. Jóvári, S. Kohara, L. Pusztai, J. Chem. Phys. 130, 064503 (2009) [2] R. Rey, J. Chem. Phys. 126, 164506 (2007)
Determining the structure of molecular liquids by combining molecular dynamics and RMC
O. Gereben1, I. Harsányi2, V. Mile2, A. Vrhovsek3, Sz. Pothoczki1, L. PUSZTAI1
References [1] R.L. McGreevy, L. Pusztai, Molecular Simulation 1 359 (1988); [2] O. Gereben, L. Pusztai, J. Comput. Chem. 33 2285 (2012); [3] V. Mile, L. Pusztai, H. Dominguez, O. Pizio, J. Phys. Chem B 113 10760 (2009)
Reverse Monte Carlo/evolutionary algorithm approach for the analysis of EXAFS data from distant coordination shells of crystalline materials
Janis TIMOSHENKO*, Andris Anspoks, Alexandr Kalinko, Alexei Kuzmin
Extended X-ray absorption fine structure (EXAFS) spectroscopy is a modern element-specific method to study the local structure of a broad class of materials [1]. The analysis of EXAFS data from the first coordination shell around the absorbing atom to obtain distributions of distances to the nearest neighbours is a well-established technique. At the same time, the experimental EXAFS data, acquired at modern X-ray sources, contain much more information, especially for crystalline materials. In this case the total EXAFS consists of contributions from significantly more distant coordination shells (up to 10 Å and further). The precise analysis of EXAFS spectra beyond the first coordination shell using conventional methods is, however, often impossible since the total number parameters, required to completely describe the local structure, is exponentially increasing with the increase of the number of coordination shells, included in the analysis [2]. This problem may be treated by reverse Monte Carlo (RMC) method [3,4]. The existing RMC implementations for EXAFS analysis, nevertheless, are rather limited due to significant computational costs of ab-initio EXAFS simulations for large atomic clusters. Therefore, in the presented study we propose another approach for the advanced analysis of EXAFS data, where we complement the conventional RMC scheme with a powerful evolutionary algorithm for a very efficient structure model optimization [5]. In this study the potentiality of the method is demonstrated on the example of temperature-dependent EXAFS study of the local structure of copper nitride.
References: [1] Rehr J J and Albers R C 2000 Rev. Mod. Phys. 72 621 [2] Provost K, Beret E, Muller D, Marcos E S and Michalowicz A 2013 J. Phys.: Conf. Ser. 430 012015 [3] McGreevy R and Pusztai L 1988 Mol. Simul. 1 359 [4] Timoshenko J, Kuzmin A and Purans J 2012 Comput. Phys. Commun. 183 1237 [5] Timoshenko J, Kuzmin A and Purans J 2014 J. Phys.: Condens. Matter 26 055401
Hydration-driven Volume Collapse in ZrW2O8: Microscopic Mechanism and Relation to Negative Thermal Expansion
Phillip TUCCIARONE
N-RMC method: Reverse Monte Carlo modeling in confined systems
Vicente SANCHEZ GIL
References: [1] V. Sánchez-Gil, E. G. Noya and E. Lomba, J. Chem. Phys. 140, 024504 (2014) [2] R. L. McGreevy and L. Pusztai, Molec. Simul. 1, 359-367 (1988)
Atomic level insights into urea induced protein unfolding
Nicola STEINKE, Christina Redfield, Christian D. Lorenz and Sylvia E. McLain
Previous work on model peptides suggests that water and hydrogen bonding interactions may have a more direct role in mediating protein folding interactions than has been previously thought. In order to probe urea's specific role in protein denaturation a model peptide, glycyl-prolyl-glycineamide(GPG), which contains a sequence which occurs in protein turns in vivo, was characterized in aqueous urea solution using neutron diffraction in combination with computational techniques. From this study, site-specific interactions between water and urea and peptide side chains can be determined. Our results indicate a direct urea-peptide bonding. Urea often replaces hydrating water around the peptide backbone.